








Servicios Personalizados
Revista

Articulo
 Artículo en XML
Artículo en XML Referencias del artículo
Referencias del artículo
 Enviar artículo por email
Enviar artículo por emailIndicadores
 Citado por SciELO
Citado por SciELO Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkArchivos de neurociencias (México, D.F.)
versión On-line ISSN 1028-5938versión impresa ISSN 0187-4705
Arch. Neurocien. (Mex., D.F.) vol.9 no.1 Ciudad de México mar. 2004
Arch Neurocien 2004; Vol. 9(1):39-46
ARTICULO DE REVISIÓN
INTERACCIÓN ENTRE LAS CÉLULAS GLIALES Y NEURONALES Y SU PAPEL EN LA MUERTE Y SOBREVIVENCIA NEURONAL
OCTAVIO GARCÍA
LOURDES MASSIEU
Departamento de Neurociencias, Instituto de Fisiología Celular,
Universidad Nacional Autónoma de México.
Correspondencia:
Octavio García.
Instituto de Fisiología Celular, UNAM.
AP. 70253, 04510 México, D.F.
E-mail: ogarcia@ifc.unam.mx
Recibido: 3 de octubre 2003
Aprobado: 23 de octubre 2003
RESUMEN
El cerebro está conformado por neuronas y células gliales, la relación entre estos dos tipos de células es fundamental para mantener la homeostasis cerebral. La excitabilidad de las células nerviosas depende en gran medida del ácido glutámico, el principal neurotransmisor excitador en el cerebro de mamíferos. La síntesis y el metabolismo del ácido glutámico, involucra una estrecha relación entre las neuronas y las células gliales. Una alteración entre los sistemas neuro-gliales glutamartérgicos puede producir la muerte de las células nerviosas a través de un mecanismo denominado excitotóxico. La muerte excitotóxica se ha asociado a la pérdida neuronal que se observa después de un episodio isquémico y en algunas enfermedades neurodegenerativas. Por otro lado, las interacciones neurogliales pueden funcionar como sistemas de neuroprotección que se activan para contrarrestar el daño excitotóxico a través de una intensa interacción metabólica que involucra la estimulación de la glucólisis y la producción de sustratos metabólicos como el piruvato y el lactato, así como la síntesis de glutatión, entre otros. En el presente trabajo se revisará el papel de los sistemas neurogliales glutamatérgicos en el daño y sobrevivencia neuronal durante la isquemia cerebral.
PALABRAS CLAVE:neuronas, glia, isquemia, glutamato.
ABSTRACT
The brain is conformed by neurons and glial cells, and the relationship between these two types of cells is fundamental in maintaining cerebral homeostasis. The excitability of nerve cells depends mainly on glutamic acid, the mayor excitatory neurotransmitter in mammalian brain. The synthesis and metabolism of glutamic acid, involves a close relationship between neurons and glial cells. An alteration of the glutamatergic neuro-glial systems can produce the death of neurons through a mechanism known as excitotoxicity. Excitotoxic death is associated with the loss of neurons observed after a cerebral ischemic episode and in some neurodegenerative diseases. On the other hand, neuro-glial systems might have a neuroprotective action to prevent excitotoxic damage through an intense metabolic interaction which involves the stimulation of glycolysis and the production metabolic substrates such as pyruvate and lactate, as well as glutathione synthesis, among others. The present work reviews the role of the glutamatergic neuro-glial system on neuronal damage and survival during cerebral ischemia.
KEY WORDS: neurons, glia, ischemia, glutamate.
El cerebro de los mamíferos está constituido principalmente por dos clases de células: las neuronas y las células gliales. Se estima que el cerebro humano contiene más de 100 mil millones de neuronas mientras que el número de células gliales supera entre 5 y 10 veces a la población neuronal1 . Durante mucho tiempo se pensó que el papel de las células gliales estaba limitado al mantenimiento de la citoarquitectura del cerebro. Sin embargo, en las últimas décadas se ha demostrado que las células gliales establecen una estrecha relación con las neuronas a través de sistemas denominados neurogliales, que se encargan de mantener la homeostasis cerebral.
Las neuronas son células altamente especializadas y morfológicamente pueden ser divididas en cuatro regiones: el cuerpo celular o soma que es el centro metabólico de la célula; las dendritas, estructuras ramificadas que reciben señales de otras células; el axón, que se extiende a partir del soma de la célula nerviosa y que conduce la información a otras neuronas, por último las terminales presinápticas que constituyen la región de la neurona que permite la comunicación con otras neuronas a través de estructuras especializadas llamadas sinápsis. Las células gliales a su vez se clasifican principalmente en tres categorías: los oligodendrocitos y las células de Schwann que forman la mielina, una membrana que sirve de barrera de aislamiento y que se encuentra involucrada en la conducción del impulso nervioso del sistema nervioso central y periférico, respectivamente; la microglia, formada por células que responden a un daño o enfermedad en el cerebro, activando una respuesta inflamatoria y fagocitando las bridas celulares o detritus, por último los astrocitos que se caracterizan por tener una forma estrellada. Los astrocitos están estrechamente vinculados con las neuronas en procesos como: la recaptura de neurotransmisores liberados durante la transmisión sináptica, el mantenimiento de la concentración de K+ y del pH, la transferencia de sustratos metabólicos y la liberación de factores tróficos que promueven el crecimiento, maduración y reparación de las células nerviosas2 . Recientemente, diversos reportes sugieren que las células gliales pueden tener otras funciones además de las ya mencionadas como son: la regulación de la excitabilidad neuronal3,4,5 , el control de la microcirculación cerebral6 y la reconstitución de los contactos sinápticos5 , lo que hace que la comunicación entre las células gliales y las neuronas sea esencial para el funcionamiento del cerebro. En este trabajo se revisará cuál es el papel de los sistemas neuro-gliales glutamatérgicos y su relación con la muerte y sobrevivencia celular asociadas con la isquemia cerebral.
La sinapsis glutamatérgica como un modelo de interacción de los sistemas neuro-gliales
La excitabilidad de las células nerviosas depende en gran medida de la acción del ácido glutámico o glutamato, que es el principal neurotransmisor excitador en el cerebro de los mamíferos. Sin embargo, bajo ciertas condiciones este aminoácido puede actuar como una potente neurotoxina. El mecanismo fisiológico y patológico de la excitabilidad neuronal involucra una estrecha comunicación entre las células gliales y las neuronas7 . La estimulación de las neuronas a través de la sinapsis glutamatérgica consiste básicamente en la despolarización de las terminales sinápticas y la liberación vesicular del glutamato de la terminal presináptica al espacio sináptico (figura 1).
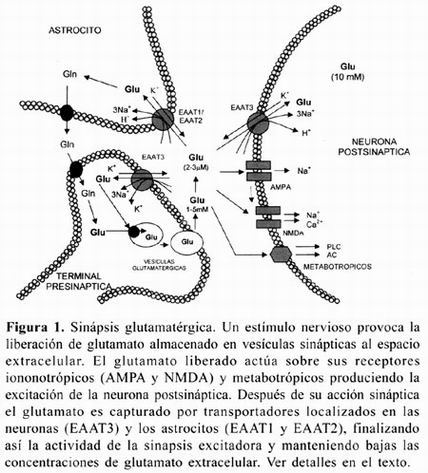
Se estima que la concentración de glutamato en las vesículas sinápticas puede alcanzar un rango de 60-210 mM y en el momento que se activa la sinapsis excitadora se pueden liberar entre 2 000 a 5 000 moléculas de glutamato alcanzando una concentración de entre 1-5 mM en el espacio sináptico8 . Las moléculas de glutamato tienen que recorrer por difusión una distancia de entre 0.5-1 μm para poder activar a receptores glutamatérgicos específicos en la membrana postsináptica (figura 1). Por último los receptores a glutamato pueden ser divididos en dos tipos: los receptores ionotrópicos y los receptores metabotrópicos. Los receptores ionotrópicos contienen un canal iónico específico para cationes y en base a la especificidad del agonista son clasificados en receptores NMDA (N-metil-D-aspatato), AMPA (α-amino-3-hidroxi-5-metil-4-isoxasolpropionato) y Kainato. La activación de los receptores glutamatérgicos en la membrana postsináptica produce un cambio en la permeabilidad iónica de la membrana y por lo tanto excitación en la neurona. Los receptores AMPA median la transmisión sináptica excitadora rápida y son permeables principalmente a Na+ . Sin embargo, el receptor a AMPA puede ser permeable al Ca2+ si está conformado por determinadas subunidades9 . Los receptores a NMDA son bloqueados fisiológicamente por Mg2+ y de manera dependiente de voltaje. Para que los receptores NMDA se activen requieren de la estimulación previa de los receptores AMPA, la despolarización producida por la activación de éstos libera el Mg2+ que se encuentra bloqueando el poro del receptor NMDA, y después la presencia de glutamato junto con la glicina hacen que el receptor entre en su estado activo lo que permite la entrada de Na+ y Ca2+ y por lo tanto la excitación de la neurona (figura 1). Por último los receptores metabotrópicos se encuentran acoplados a proteínas G y modulan la producción de mensajeros intracelulares por la activación de la fosfolipasa C (PLC) o de la adenilato ciclasa (AC) (figura 1). por último la concentración intracelular del glutamato en el cerebro es de aproximadamente 10 mM mientras que la concentración extracelular es extremadamente baja cerca de 2-3 μM. CEn lo presedente anteriormente cuando la sinapsis excitadora se activa la concentración de glutamato en el espacio sináptico puede alcanzar una concentración de 1-5 mγ8 , si la concentración de glutamato no disminuye después de ser liberado este neurotransmisor puede resultar tóxico a las neuronas incluso a concentraciones de 10 μM, si permanece por un tiempo prolongado en el medio extracelular10 . Las concentraciones extracelulares de glutamato se regulan por la acción de sistemas de captura de alta afinidad dependientes de Na+ localizados en neuronas y células gliales (figura1). Después de actuar sobre sus receptores, el glutamato es eliminado rápidamente del espacio sináptico en un tiempo aproximado de 50-200 μs8 finalizando así la actividad de la sinápsis excitadora. La captura de glutamato se lleva acabo principalmente por los astrocitos, esta captura es mayor a la de las propias neuronas glutamatérgicas por lo que se considera que la activación de los transportadores localizados en los astrocitos, es el mecanismo predominante utilizado en los sistemas neuro-gliales para mantener bajas las concentraciones extracelulares de glutamato en el cerebro11.
En la actualidad se han clonado 5 tipos de transportadores a glutamato, los predominantemente gliales conocidos como GLAST y GLT-1 clonados originalmente del cerebro de rata, y sus homólogos humanos EAAT1 y EAAT2, respectivamente. El transportador EAAC1 y su homólogo en humanos EAAT3 que se localiza principalmente en las células neuronales, el transportador EAAT4 expresado en células cerebelares de Purkinje, y finalmente el transportador EAAT5 expresado en retina. En los últimos años se ha demostrado que tanto en neuronas como en células gliales existe una expresión diferencial de los transportadores e incluso una colocalización de los diferentes subtipos de transportadores a glutamato en las células 12
La captura de glutamato estimula la glucólisis
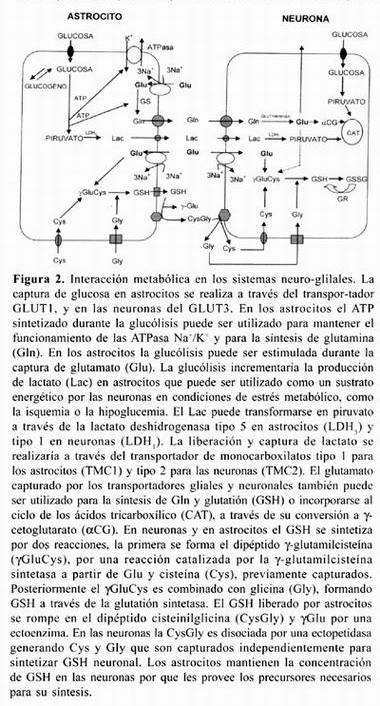
La captura de glutamato por los transportadores requiere de energía. El costo energético de esta captura requiere de 1 molécula de ATP por molécula de glutamato transportada asociada a 3 iones de Na+ 12 . El ATP requerido para el transporte de glutamato es obtenido por medio de la oxidación de la glucosa capturada por transportadores específicos ubicados en los astrocitos. Durante el proceso glucolítico se producen dos ATP, un ATP es usado para la expulsión de los iones Na+ por la ATPasa Na+ /K+ y el otro ATP es utilizado para la síntesis de glutamina, además de producirse dos moléculas de lactato por cada molécula de glucosa. Se ha propuesto que la activación fisiológica de la sinapsis glutamatérgica podría estimular la glucólisis de los astrocitos13 a través del siguiente mecanismo: una vez liberado el glutamato de las terminales presinápticas el neurotransmisor sería capturado principalmente por los transportadores localizados en los astrocitos. Como el glutamato es cotransportado con iones Na+ causaría un incremento en la concentración intracelular de Na+ en los astrocitos lo que conduciría a la activación de la ATPasa Na+ /K+ (figura 2). La activación de la ATPasa Na+ /K+ estimularía la glucólisis13 . Adicionalmente el glutamato que es capturado por los astrocitos puede ser metabolizado por diversas vías y ser utilizado en la formación de glutamina o entrar en el ciclo del ácido tricarboxílico y servir como una fuente energética (figura 2). La formación de glutamina es catalizada por la glutamina sintetasa una enzima localizada en los astrocitos y en menor cantidad en los oligodendrocitos, pero ausente en las neuronas. El metabolismo oxidativo del glutamato es iniciado por su conversión a γ-cetoglutarato, mismo que es utilizado en el ciclo del ácido tricarboxílico; esto puede ser mediado por la glutamato deshidrogenasa o por transaminación11.
Metabolismo del glutatión en los sistemas neuro-gliales
Comparado con otros órganos del cuerpo, el cerebro es el órgano más susceptible al daño oxidativo debido a diversos factores que incluyen la alta utilización del oxígeno, la presencia de un exceso de ácidos grasos insaturados que son blanco de la peroxidación lipídica, la baja o moderada actividad de enzimas antioxidantes como la superóxido dismutasa (SOD) y la catalasa, y la presencia de grandes concentraciones de hierro que favorecen la formación de especies reactivas de oxígeno (ERO). Las ERO son especies químicas de oxígeno que presentan uno o más electrones desapareados en su último orbital, esta inestabilidad los vuelve reactivos pudiendo alterar la estructura molecular de lípidos, proteínas y ácidos nucléicos. La generación de ERO es rápida y constante en el cerebro, ya que este órgano utiliza cerca del 20% del oxígeno consumido por el cuerpo, pero solo constituye un 2% de su peso, lo que indica que en el cerebro existe un gran metabolismo oxidativo. Por lo tanto, la detoxificación de las ERO es esencial en el cerebro para evitar el daño oxidativo.
El tripéptido glutatión (GSH, γ-L-glutamil-cisteinil-glicina), es un sistema muy importante para la defensa celular contra las ERO. En el cerebro la concentración de GSH tiene un rango de aproximadamente 1 a 3 mM. En particular, la concentración de GSH en los astrocitos es de 16-50 nmol/mg proteína y es mayor que en las células neuronales14 . El GSH reacciona directamente contra los radicales libres a través de reacciones enzimáticas y no enzimáticas. En las reacciones no enzimáticas el GSH reacciona con radicales tales como el anión superóxido, el óxido nítrico o el radical hidroxil, mientras que en las reacciones enzimáticas el GSH es el donador de electrones para la reducción de peróxidos en una reacción catalizada por la glutatión peroxidasa (GPx). El producto final de la oxidación del GSH es el glutatión disulfido (GSSG). El GSH es regenerado del GSSG por la reacción catalizada por la glutatión reductasa (GR)15
El GSH es sintetizado por la acción consecutiva de dos enzimas, la γ-glutamilcisteina sintetasa que utiliza al glutamato y a la cisteína como sustratos formando el dipéptido γ-glutamilcisteina (γGluCys). Posteriormente el γGluCys es combinado con la glicina para generar GSH por medio de una reacción catalizada por la glutatión sintetasa (figura 2). El contenido de glutatión en las células nerviosas depende fuertemente de la disponibilidad de sus precursores. En el cerebro los astrocitos aportan a las neuronas precursores necesarios para la síntesis de GSH por lo que se establece una interacción metabólica en los sistemas neuro-gliales. Se propone que el GSH es previamente sintetizado en los astrocitos y liberado de estos e hidrolizado por una ectoenzima conocida como γ-glutamiltranspeptidasa (γGT), localizada en la membrana plasmática de los astrocitos. En esta reacción se produce el dipéptido cisteinilglicina (CysGly) y γ-Glu. El CysGly es disociado por una ectopeptidasa localizada en las neuronas generando cisteína y glicina que son capturados de manera independiente por las neuronas y utilizados para volver a sintetizar GSH (figura 2). La glutamina liberada de los astrocitos es usada por las neuronas como un precursor del glutamato necesario como neurotransmisor y para la síntesis de GSH14 (figura 2). Sin embargo, en las neuronas la disponibilidad de la cisteína es el paso limitante para la formación de GSH. Recientemente algunos trabajos han reportado que la captura de cisteína necesaria para la síntesis de GSH, está asociada a la captura de glutamato a través de los transportadores neuronales16,17.
La muerte excitotóxica y los sistemas neuro-gliales
Como mencionamos anteriormente un incremento en la concentración extracelular de glutamato puede resultar tóxico para las células nerviosas. El efecto tóxico del glutamato se conoce desde finales de la década de los 50's del siglo pasado cuando Lucas y Newhouse18 , demostraron que la administración del glutamato y análogos de este aminoácido producían degeneración en las células nerviosas de la retina. Posteriormente Olney19 , propuso que la muerte neuronal inducida por este aminoácido se debía a una despolarización prolongada de las neuronas, acuñando el término excitotoxicidad para referirse al daño celular producido por la actividad prolongada de los receptores glutamatérgicos. En particular, la activación del receptor NMDA ha sido la vía mejor caracterizada de la muerte excitotóxica. Se propone que la excesiva estimulación del receptor NMDA, conduce a una entrada masiva de Ca2+ a la célula activando una variedad de enzimas celulares dependientes de este ión tales como proteasas, las cuales contribuirían al daño celular al degradar proteínas estructurales; fosfolipasas contribuyendo a la ruptura de la membrana celular; endonucleasas, generando alteraciones en el DNA y la activación de la sintasa de óxido nítrico (NOS), enzima involucrada en la generación de óxido nítrico (NO), y una subsecuente producción de ERO20 (figura 3). Recientemente se ha propuesto que la mitocondria podría tener un papel preponderante en la muerte excitotóxica21 . Diversas evidencias experimentales sugieren que la muerte excitotóxica es un evento común que se presenta en algunos desórdenes neurológicos como la isquemia cerebral, la hipoglicemia y la epilepsia, y en algunas enfermedades neurodegenerativas como la de Huntington, Alzheimer y la de Parkinson22.
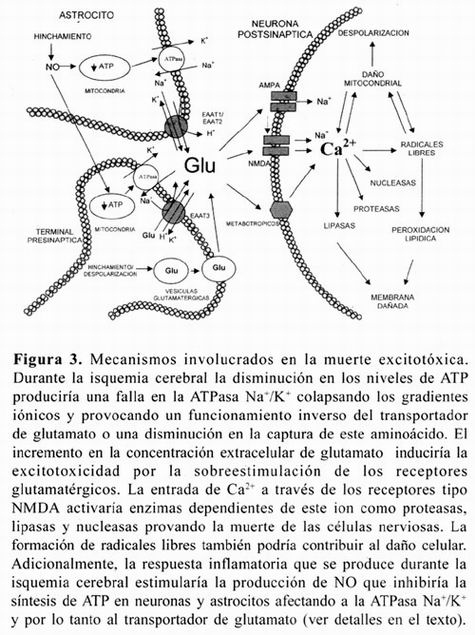
Sin embargo, en todas estas condiciones se desconoce cuál es el origen del glutamato implicada en la muerte excitotóxica. Algunos estudios sugieren que una alteración de los transportadores encargados remover el glutamato del espacio sináptico podrían favorecer el incremento en la concentración extracelular de este neurotransmisor y con ello la muerte excitotóxica. Esta alteración ocurriría subsecuentemente a la deficiencia energética por ejemplo, durante la isquemia cerebral la drástica disminución en los niveles de ATP podría generar un mal funcionamiento de la ATPasa Na+ /K+ . Esto provocaría el colapso de los gradientes iónicos afectando directamente la actividad de los transportadores de glutamato que son dependientes de Na+ , produciendo su funcionamiento inverso, o una deficiencia en la captura de este aminoácido, tanto en neuronas como en células gliales23 . La presencia de NO también podría influir en la función inversa de los transportadores y producir un incremento de la concentración extracelular de glutamato durante la isquemia. Este mecanismo está relacionado con la respuesta inflamatoria que se observa después del daño cerebrovascular la cual estimularía la producción de NO en los astrocitos. El NO podría difundir a las neuronas e inhibir su respiración y disminuir los niveles de ATP, lo cual favorecería la liberación de glutamato al espacio extracelular a través de un funcionamiento inverso del transportador. Esto podría ocurrir también en los astrocitos. El glutamato liberado por la glia y las neuronas estimularía a los receptores glutamatérgicos de las neuronas vecinas y produciría una muerte excitotóxica24 . Por otro lado, en experimentos con ratones knock out de los transportadores gliales GLAST y GLT-1, se observó un incremento en los niveles extracelulares de glutamato y la presencia de muerte excitotóxica, mientras que en los animales knock out del transportador neuronal EAAC1 no se presentaba neurodegeneración25 . A su vez, la inhibición de la expresión de los transportadores gliales favorece la pérdida neuronal y la generación de crisis epilépticas26 , y exacerba el daño neuronal isquémico27
La interacción neuro-glial durante la isquemia cerebral
Durante un proceso patológico como la isquemia cerebral, los sistemas neuro-gliales pueden ser afectados provocando un desajuste en la homeóstasis cerebral que puede conducir a la muerte celular. Sin embargo, la duración del periodo isquémico puede determinar si el daño celular ocurre en las neuronas o en los astrocitos. Por ejemplo, en la isquemia generada por la oclusión de 4 vasos cerebrales se ha observado que un periodo de 2 ó 3 minutos de isquemia es suficiente para producir daño neuronal mientras que los astrocitos sufren daño después un periodo isquémico de 30 a 60 minutos. Estos resultados son similares a los observados en la isquemia generada por la oclusión de arteria cerebral media. En este modelo se ha reportado que un periodo isquémico de 40 minutos es suficiente para producir muerte neuronal mientras que los astrocitos requieren un periodo isquémico de 360 minutos28 . En cultivos celulares expuestos a privación de glucosa y oxígeno (DGO), condiciones utilizadas como modelo de isquemia in vitro , durante un periodo de 45-60 minutos puede ser letal para las neuronas pero no para los astrocitos. La pérdida neuronal y la sobrevivencia de los astrocitos estarían asociadas con la capacidad de las células para mantener el potencial de membrana mitocondrial y los niveles de ATP, además de que se ha sugerido una producción diferencial de ERO, ya que en las neuronas se produce preferentemente superóxido (O 2 .- ) y en los astrocitos se produce NO29. El hecho de que los astrocitos puedan tolerar con mayor eficiencia un periodo isquémico puede es-tar relacionado con su actividad metabólica2, con la eficacia para capturar glutamato11 y con un menor número de receptores glutamatérgicos tipo NMDA30. Sin embargo, algunos trabajos han demostrado que las células gliales pueden sufrir daño excitotóxico cuando son expuestas a altas concentraciones de glutamato (1-10 mM) por periodos prolongados (de 16-24 horas) 31
Por otro lado, el glucógeno es una gran reserva de energía y en el cerebro se encuentra selectivamente localizado en los astrocitos. Bajo condiciones de privación energética es utilizado para mantener los niveles de ATP. Los astrocitos de corteza de rata expuestos a una DGO pueden metabolizar el glucógeno y utilizar la glucólisis anaerobia, lo que incrementa la producción de lactato 32 . Sin embargo, la captura de glucosa en astrocitos también se podría incrementar bajo condiciones isquémicas como un mecanismo que permitiría generar ATP y conservar la actividad de la ATPasa Na+ /K+ . Así se mantendría el funcionamiento normal del transportador de glutamato evitando la muerte excitotóxica en las neuronas, al ser éste removido 13,33 . El incremento en la actividad glucolítica en los astrocitos podría generar un incremento en la producción de lactato que sería utilizado como un sustrato metabólico por las neuronas en condiciones de deficiencia energética como el caso de la isquemia y la hipoglucemia33,34 . Esta hipótesis es apoyada por diversas evidencias obtenidas de experimentos in vivo e in vitro que demuestran: 1. que existe un incremento en la concentración de lactato durante la isquemia cerebral35, 2. que el lactato es altamente oxidado por las neuronas36 y 3. que el lactato mantiene los niveles de ATP y promueve la sobrevivencia y actividad neuronal cuando los niveles de glucosa son bajos32,35 . En ausencia de un sustrato metabólico adecuado, las neuronas pueden transformar el lactato en piruvato y posteriormente incorporarlo al ciclo de los ácidos tricarboxílicos vía la formación de acetil CoA generando 18 ATP por molécula de lactato32 . También se ha propuesto que algunos de los carbones de la molécula de lactato puedan incorporarse a pozas de aminoácidos tales como los neurotransmisores glutamato, aspartato y GABA2 . Además, la existencia de transportadores específicos para lactato en astrocitos y neuronas apoya la existencia un mecanismo coordinado en ambas células37 . Sin embargo, es posible que los astrocitos no solo liberen lactato sino también piruvato38 . El piruvato liberado por los astrocitos podría proteger a las neuronas de un daño tóxico no actuando sólo como un sustrato energético sino también como un atrapador de radicales libres38,39 . Esta idea es apoyada en diversos trabajos que demuestran que el piruvato puede funcionar como un excelente compuesto neuroprotector38-44.
Durante la isquemia cerebral los sistemas neurogliales también pueden ser afectados por daño oxidativo subsecuente a la reperfusión posisquemia. Por otro lado, existen reportes donde se demuestra que animales que tienen disminuidos los niveles de GSH son más susceptibles al daño isquémico45 , y que durante la isquemia cerebral existe una elevada concentración extracelular de GSH. Este último fenómeno puede ser el resultado de la capacidad de las células del cerebro, en particular de los astrocitos, para liberar este tripéptido 46 . El GSH es un antioxidante muy importante contra la toxicidad del peróxido de hidrógeno (H2O2 ). El H2O2 es un peróxido que se genera en altas concentraciones en el cerebro y que podría estar involucrado en los procesos neurodegenerativos asociados con la enfermedad de Parkinson, o contribuir al desarrollo de la enfermedad de Alzheimer47,48 . La depleción de GSH es uno de los primeros indicadores de estrés oxidativo durante la progresión de la enfermedad de Parkinson y al parecer la depleción de GSH es paralela a la severidad de la enfermedad49 . En algunos reportes se propone que un solo astrocito podría proteger a 20 neuronas contra la toxicidad del H2O2 50 y que la depleción en los niveles de GSH podría estar involucrada a la alteración del funcionamiento de los transportadores de glutamato, afectando directamente la captura y concentración de los sustratos necesarios para la síntesis de GSH51,52 . Por otro lado, la producción de NAD(P)H necesaria para la formación de GSH se obtiene principalmente a través de la vía de las pentosas fosfato. Se ha demostrado que en astrocitos el flujo de la vía de las pentosas fosfato es mayor con respecto a las neuronas y se incrementa hasta tres veces durante una exposición de H2O2 53,54.
Como mencionamos anteriormente los astrocitos están involucrados en la síntesis de GSH neuronal ayudando a estas células a protegerse contra un daño oxidativo. Sin embargo, los astrocitos también podrían ser más tolerantes al daño oxidativo porque presentan una mayor actividad de enzimas antioxidantes como la catalasa, y una concentración 2.5 veces mas alta de vitamina E que las neuronas, la cual es necesaria para evitar la peroxidación lipídica. La pérdida o alteración de las defensas antioxidantes en los sistemas neurogliales también podría contribuir a la disminución de los niveles de ATP que se observa durante la isquemia cerebral, pues algunas ERO pueden inhibir la actividad de diversos complejos de la cadena de transporte de electrones (CTE) mitocondrial. Por ejemplo durante la DGO la disminución de los niveles de ATP podría estar asociada a la inhibición del complejo I en las neuronas mientras que los astrocitos no muestran disminución de la actividad de la CTE mitocondrial, y tampoco una disminución en los niveles de ATP54 . La inhibición de los complejos de la CTE mitocondriales en las neuronas correlaciona con una mayor daño celular mientras que los astrocitos al no presentar esta inhibición pueden tolerar la presencia de ERO55 . Adicionalmente, el NO producido en la glia durante una respuesta inflamatoria, se podría difundir a otras neuronas y afectar sus mitocondrias produciendo una disminución en los niveles de ATP, ya sea directamente o a través de la formación de peroxinitrito (ONOO-)24,55.
CONCLUSIÓN
Las interacciones entre neuronas y astrocitos son necesarias para mantener la homeostasis en el cerebro ya que existe una estrecha relación entre ambos tipos de células. La sobrevivencia y muerte celular que se presenta en algunas enfermedades neurodegenerativas o durante la isquemia cerebral podría estar vinculada no sólo a las características particulares de cada tipo celular, sino también a la interacción física, metabólica y fisiológica que existe entre estas células.
AGRADECIMIENTOS
Este trabajo fue realizado gracias al apoyo IN222503 (PAPIIT) a Lourdes Massieu y una beca de CONACYT 163330 y DGEP-UNAM a Octavio García.
REFERENCIAS
1. Kandel ER. The nervous system has two classes of cells. En Kandel ER, Schwartz JH, Jessell TM, editores. Principles of neuronal science. USA: McGraw-Hill, 2000;19-35. [ Links ]
2. Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci 1996; 16:877-85. [ Links ]
3. Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science 2002; 298:556-62. [ Links ]
4. Auld DS, Robitaille R. 2003. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron 2003;40:389-400. [ Links ]
5. Newman EA. 2003. New roles for astrocytes: regulation of synaptic transmission. Trends Neurosci 2003; 26:536-42. [ Links ]
6. Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 2003;6:43-50. [ Links ]
7. Nedergaard M, Takano T, Hansen AJ. Beyond the role of glutamate as neurotransmitter. nat rev neurosci 2002;3:748- 55. [ Links ]
8. Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci 1996; 19:163-71. [ Links ]
9. Pellegrini-Giampietro DE, Gorter JA, Bennett MVL, Zukin SR. The GluR2 (GluR-B) hypothesis: Ca 2+ permeable AMPA receptor in neurological disorders. Trends Neurosci 1997; 20:464-70. [ Links ]
10. Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacol 1998;37: 1419-29. [ Links ]
11. Hertz L, Dringen R, Schousboe A, Robinson SR. Astrocytes: glutamate producers for neurons. J Neurosci Res 1999; 57:417-28. [ Links ]
12. Danbolt NC. Glutamate uptake. Prog Neurobiol 2001;65:1-105 [ Links ]
13. Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 1994; 91:10625-9. [ Links ]
14. Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 2000; 267:4912-6. [ Links ]
15. Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol 2000; 62:649-71. [ Links ]
16. Himi T, Ikeda M, Yasuhara T, Nishida M, Morita I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J Neural Trans 2003; 110: 1337-48. [ Links ]
17. Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J Neurochem 2003;84:1332-39. [ Links ]
18. Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. Arch Ophtal 1957; 58:193-201. [ Links ]
19. Olney JW. Glutamate-induced retinal degeneration in neonatal mice. Electron microscopy of the acutely evolving lesion. J Neuropathol Exp Neurol 1969; 28:455-74. [ Links ]
20. Bittigau P, Ikonomidou C. Glutamate in neurological diseases. J Child Neurol 1997; 12:471-485. [ Links ]
21. Nicholls DG, Budd SL. Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta 1998;1366:97-112. [ Links ]
22. Obrenovitch TP, Urenjak J. Altered glutamatergic transmission in neurological disorders: from high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol 1997; 51:39-87. [ Links ]
23. Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 2000; 403:316-21. [ Links ]
24. Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 2001; 21:6480-91. [ Links ]
25. Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA Jin L, Kunel RW, Kanai Y, Hediger MA, Wang Y Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996;16:675-86. [ Links ]
26. Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 1997;276:1699-702. [ Links ]
27. Rao VL, Dogan A, Todd KG, Bowen KK, Kim BT, Rothstein JD, Dempsey RJ. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Neurosci 2001; 21:1876-83. [ Links ]
28. Fern R. Ischemia: astrocytes show their sensitive side. Prog Brain Res 2001;132:405-11. [ Links ]
29. Reichert SA, Kin-Han JS, Dugan LL. The mitochondrial permeability transition pore and nitric oxide synthase mediate early mitochondrial depolarization in astrocytes during oxygen-glucose deprivation. J Neurosci 2001;21:6608-16. [ Links ]
30. Seifert G, Steinhäuser C. Ionotropic glutamate receptors in astrocytes. Prog Brain Res 2001;132:287-99. [ Links ]
31. Chen C-J, Liao S-L, Kuo J-S. Gliotoxic action of glutamate on cultured astrocyte. J Neurochem 2000;75:1557-65. [ Links ]
32. Dienel GA, Hertz L. Glucose and lactate metabolism during brain activation. J Neurosci Res 2001;66:824-38. [ Links ]
33. Stanimirovic DB, Ball R, Durkin JP. Stimulation of glutamate uptake and Na, K-ATPase activity in rat astrocytes exposed to ischemia-like insults. Glia 1997;19:123-34. [ Links ]
34. Voutsinos-Porche B, Bonvento G, Tanaka K, Steiner P, Welker E, Chatton J-Y, Magistretti PJ, Pellerin L. Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 2003;37:275-86. [ Links ]
35. Smith D, Pernet A, Hallett WA, Bingham E, Marsden PK, Amiel SA. Lactate: a preferred fuel for human brain metabolism in vivo. J Cereb Blood Flow Metab 2003;23:658-64. [ Links ]
36. Bouzier-Sore A-K, Merle M, Magistretti PJ, Pellerin L. Feeding active neurons: (re)emergence of a nursing role for astrocytes. J Physiol (Paris) 2002;96:273-82. [ Links ]
37. Pellerin L. Lactate as a pivotal element in neuro-glia metabolic cooperation. Neurochem Int 2003;43:331-8. [ Links ]
38. Wang XF, Cynader MS. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci 2001;21:3322-31. [ Links ]
39. Desagher S, Glowinski J, Premont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci 1997; 17:9060-7. [ Links ]
40. Ruiz F, Alvarez G, Pereira R, Hernandez M, Villalba M, Cruz F, Cerdan S, Bogonez E, Satrustegui J. Protection by pyruvate and malate against glutamate-mediated neurotoxicity. Neuroreport 1998;9:1277-82. [ Links ]
41. Maus M, Marin P, Israel M, Glowinsky J, Premont J. Pyruvate and lactate protect striatal neurons against N-methyl-D asapar-tate-induced neurotoxicity. Eur J Neurosci 1999;11:3215-24. [ Links ]
42. Lee JY, Kim YH, Koh JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci 2001;21 (RC171):1-6. [ Links ]
43. Arias C, Montiel T, Quiroz-Baez R, Massieu L. ß-Amyloid neurotoxicity is exacerbated during glycolysis inhibition and mitochondrial impairment in the rat hippocampus in vivo and in isolated nerve terminals: implications for Alzheimer's disease. Exp Neurol 2002; 176:163-74. [ Links ]
44. García O, Massieu, L. Glutamate uptake inhibitor L-trans-pyrrolidine 2,4-dicarboxylate becomes neurotoxic in the presence of subthreshold concentrations of mitochondrial toxin 3-nitropropionate: involvement of mitochondrial reducing activity and ATP production. J Neurosci Res 2003;74:956-66. [ Links ]
45. Mizui T, Kinouchi H, Chan PH. Depletion of brain gluthathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am J. Physiol. 1992;262:H313-7. [ Links ]
46. Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanims. J Neurochem 2001; 77:1601-10. [ Links ]
47. Behl C. Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid ß protein toxicity. Cell 1994; 77:817-827. [ Links ]
48. Bharath S, Hsu M, Kaur D, Rajagopalan S, Andersen JK. Glutathione, iron and Parkinson´s disease. Biochem Pharmacol 2002;64:1037-48. [ Links ]
49. Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur J Biochem 2000; 267:4904-11. [ Links ]
50. Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci 1996; 16:2553-2562. [ Links ]
51. Himi T, Ikeda M, Yasuhara T, Murota SI. Oxidative neuronal death caused by glutamate uptake inhibition in cultured hippocampal neurons. J Neurosci Res 2003; 71:679-88. [ Links ]
52. Ré DB, Boucraut J, Samuel D, Birman S, Kerkerian-Le Goff L, Had-Aissouni L. Glutamate transport alteration triggers differentiation-state selective oxidative death of cultured astrocytes: a mechanism different from excitotoxicity depending on intracellular GSH contents. J Neurochem 2003;85:1159-70 [ Links ]
53. Ben-Yoseph O, Boxer PA, Ross BD. Assessment of the role of the glutathione and pentose phosphate pathways in the protection of primary cerebrocortical cultures from oxidative stress. J Neurochem 1996;66:2329-37. [ Links ]
54. Almeida A, Delgado-Esteban M, Bolaños JP, Medina JM. Oxygen and glucose deprivation induce mitocondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem 2002;81:207-17. [ Links ]
55. Bolaños JP, Heales SJR, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem 1995; 64:1965-72. [ Links ]